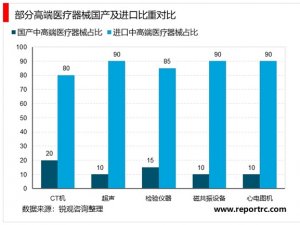
医疗器械临床试验相关规定梳理
医疗器械临床试验是指在经资质认定的医疗器械临床试验机构中,对拟申请注册的医疗器械在正常使用条件下的安全性和有效性进行确认或验证的过程。由于器械与药品之间差异巨大,其临床试验与药物临床试验存在诸多不同:
(1)产品性能
不同领域医疗器械产品跨度大:不同治疗领域药品的临床试验基本相同或相似,但对于医疗器械来说,不同领域的器械,几乎就是完全不同的操作流程,例如植入性器械和体外诊断试剂,试验的目标群体和执行人员完全不同;眼科、齿科和大型影像设备的方案设计和评估指标则没有可比性;有源和无源的医疗器械,遵循的标准分属两个体系。
医疗器械产品有效性一定程度上依赖于研究者的操作:药物经由不同的给药途径进入血液循环,在此过程中研究者的操作几乎不会影响到药物的有效性。但对于
许多医疗器械(尤其是植入性器械)来说,操作过程对产品有效性影响很大。如果手术操作上存在某些问题而又没被发现,最终很可能得到相反的试验结果,并且不同的研究者进行操作,结果也许会截然不同。所以临床试验监查员在监查过程中,需要具备一定的手术知识,根据手术记录和操作发现潜在的问题,并及时与研究者沟通交流。
医疗器械产品更新换代较快:药品的生命周期往往可以达到10-20年甚至更长;而医疗器械则一般以2-5年为一个周期。很多情况下,这一代的产品的临床试验刚刚结束,下一代产品就已经推出,而这会直接影响到公司对临床试验的期望值和整体战略安排。
(2)试验设计
药物通常更容易进行对照、随机、双盲等复杂的试验方案设计,药品剂量、给药方式调整等也相对容易实现。药物临床试验的疗效、安全性评价指标相对成熟,有公认的标准可供参考。同时,中心实验室、适用于临床试验数据采集和传输的EDC(ElectronicDataCaptureSystem)系统、为临床试验中随机化分配、受试者管理、药品管理等服务的中央化随机系统IVRS/IWRS(InteractiveVoiceResponseSystem/InteractiveWebResponseSystem)也相对比较完善。
然而,不同的医疗器械试验设计差异巨大。由于条件限制,创新型器械往往难以找到合适的对照品。如果器械的使用涉及手术操作,也基本无法实现双盲,往往采用假手术对照(ShamControl)来进行效果的考察。此外,医疗器械的疗效、安全性指标没有公认的标准,在观察指标的选择上存在难度,中心实验室、EDC等试验系统也与药物存在一定差距。
样本量
药物临床试验中不同时期的样本量规定有所不同,一般期数越高,所需的样本量越多。根据2002年12月SFDA发布的《药品注册管理办法》规定:药物临床研究的受试例数应当根据临床研究的目的,符合相关统计学的要求和办法所规定的最低临床研究病例数要求,最低临床试验病例数分别为:I期试验20-30例,Ⅱ期100例,Ⅲ期300例,Ⅳ期2000例。而由于医疗器械存在产品范围跨度大,各类
产品标准无法统一等特点,因此并没有像药品临床试验有统一要求。曾经在1997年发布的《医疗器械产品临床验证暂行规定》中对于不同类型产品的临床验证期限和病例数有相应的规定。
表:不同医疗器械产品的临床验证期限和病例数规定
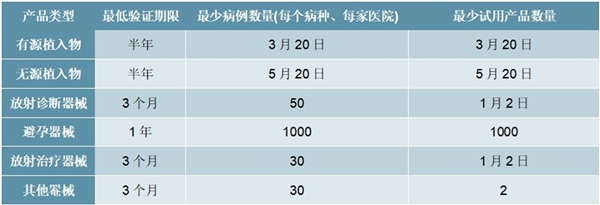
资料来源:《医疗器械产品临床验证暂行规定》(1997,已废止),锐观咨询整理
但由于科技的飞速发展,越来越多新型医疗器械被研发出来,各产品之间性能差异加大,难以用这样简单的划分使所有医疗器械都能相匹配。因此,现在已经取消了对临床试验时间以及病例数的要求,医疗器械临床试验方案一般都是由企业和试验医疗单位共同研究确定。《医疗器械临床试验设计指导原则》规定,为实现样本(受试人群)代替总体(目标人群)的目的,临床试验需要一定的受试者数量(样本量)。样本量大小与主要评价指标的变异度呈正相关,与主要评价指标的组间差异呈负相关。样本量一般以临床试验的主要评价指标进行估算。需在临床试验方案中说明样本量估算的相关要素及其确定依据、样本量的具体计算方法。
体外诊断试剂的临床试验则有明确的样本量要求,《体外诊断试剂临床试验技术指导原则》规定,第二类产品临床试验的总样本数至少为200例,第三类产品由于可能对人体具有潜在危险,必须对其安全性和有效性进行严格的控制,故临床试验的总样本数至少为1000例。对于一些特殊产品,临床试验的总样本数有特定要求。
表:特殊要求诊断试剂的临床试验总样本数要求
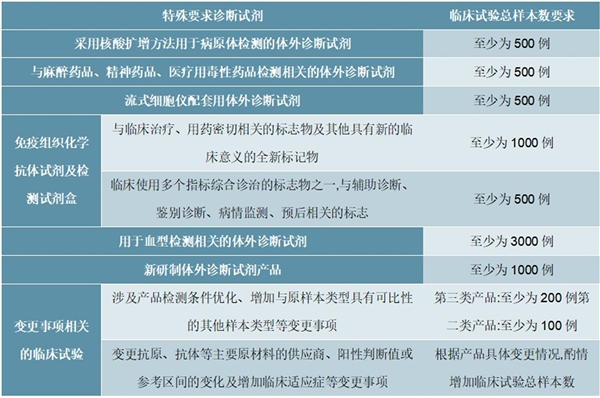
资料来源:《体外诊断试剂临床试验技术指导原则》,锐观咨询整理
同时,还要求第三类体外诊断试剂申请人应当选定不少于3家(含3家)、第二类体外诊断试剂申请人应当选定不少于2家(含2家)临床试验机构,按照有关规定开展临床试验。
(3)、试验分期
与药物临床试验通常需要划分Ⅰ、Ⅱ、Ⅲ、Ⅳ期不同,医疗器械临床试验一般不进行分期或没有明确的分期,只需单次试验来达到临床试用或临床验证的目的。原先的法规规定医疗器械临床试验分医疗器械临床试用和医疗器械临床验证,但新的《医疗器械临床试验质量管理规范》取消了这样的划分,只规定对于未在境内外批准上市的新产品,安全性以及性能尚未经医学证实的,为了充分保护受试者权益,临床试验方案设计时应当先进行小样本可行性试验,而后根据情况方可开展较大样本的安全有效性试验。此外,对于特殊的植入性器械或创新型器械等,则可选择先进行多次小规模的预实验,初步观察结果后再适当扩大试验规模至规定范围内即可。
(4)、试验周期
由于药物临床试验涉及分期,总体的周期较长,大约在3-10年及以上,而医疗器械的临床试验周期相对较短,一般仅需要半年到3年左右。资金投入
临床研究是医药创新产业的关键环节。其中,临床试验是验证药物在人体内安全性和有效性的唯一方法。从全球经验来看,单个药物临床试验从启动到完成一般需要4-6年,平均成本超过10亿元人民币,时间和资金投入在整个新药研发中约占70%。KMR集团在2016年进行了一项新药临床试验成本研究,收集了2016年收入在TOP20之列的7家大型制药公司在2010-2015年间开展的726项介入性研究。从临床方案批准到最终临床报告公布,临床试验各期花费的中位值分别是:I期340万美元、II期860万美元、III期2140万美元。通过这些数据可见新药研发临床试验成本之高。
而对于医疗器械来说,由于其产品种类跨度大,也没有统一的临床试验标准,临床试验方案需要根据不同的医疗器械分别研究制定。因此,在医疗器械临床试验上的成本也跨度较大,从几百万到上千万均有。根据不同医疗器械临床试验实施难度的不同,所花费的资金也不一样。像第三类医疗器械,一般需要植入人体,具有较高风险,其临床试验实施难度大,所以需要投入的成本也更多。
(5)、申办方的认知
药物临床试验是研发的重要组成部分,在每年进行的大量药物临床试验中,不仅有支持药品注册的临床试验,还包括了大量的上市后研究、探索性研究、流行病学调查等,药企很了解临床试验为企业带来的长远影响。
对于医疗器械而言,临床试验主要是一个验证的过程,重要性并没有药品那么高。
由于产品周期短,导致申办方不愿在临床试验中花费过多时间,故大部分临床试验为支持器械注册而生。产品性质和周期决定了器械临床试验很难具有像药品临床试验那样对于企业的重要战略地位。
(6)、试验记录保存
根据《药物临床试验质量管理规范》第八章第五十二条规定,研究者和申办者应保存药物临床试验资料至临床试验终止后5年,而医疗器械的临床试验记录则需要在试验结束后保留至少10年。